Short-read Methylation Pipeline Using Nextflow
Giang Nguyen
04 May 2026
We evaluated both BSBolt and Rastair for methylation calling. BSBolt provides accurate and fast bisulfite sequencing alignments and methylation calls, outperforming Bismark, BSSeeker2, BISCUIT, and BWA-Meth based on alignment accuracy and methylation calling accuracy. Rastair achieves F1 scores exceeding 0.99 for datasets above 30x depth while processing a 30x depth file in under 30 minutes with 32 CPU cores.
References:
nf-core/methylation is widely regarded as a leading pipeline for methylation calling for short-read sequencing, backed by a large and active community. However, it does come with several limitations:
- Reputation bias: Widely adopted tools are often retained as defaults, even when they may no longer offer the best performance. This can lead to unnecessary resource consumption and suboptimal efficiency in certain steps.
- Legacy features: Some integrated tools have not been actively maintained for years, resulting in performance gaps. Despite this, they are often preserved for backward compatibility, increasing codebase complexity and the maintenance burden when introducing new features.
- Customization constraints: The pipeline is designed for general-purpose and research-oriented use. In industrial production settings, workflows typically need to be streamlined—retaining only essential steps and achieving near-linear scalability with resources. In such cases, a more minimal and purpose-built approach is often more effective.
Therefore, we developed the nf-short-read-methylation specifically to support large-scale methylation analysis from bisulfite sequencing data (BSBolt) and TAPs sequencing data (Rastair).
Architecture
Key Features
- Two Methylation Pathways: BSBolt (traditional bisulfite) and Rastair (TAPS-based)
- Multiple Input Formats: FASTQ (full pipeline), BAM, and CRAM (skip alignment, auto-convert)
- Quality Control: Fastp
- M-bias Analysis: Automatic M-bias calculation and trimming optimization
- Flexible Deduplication: Samtools MarkDup (BSBolt), GATK MarkDuplicates (Rastair)
- Cross-sample Aggregation: Methylation matrix generation for comparative analysis
- CRAM Compression: Built-in CRAM→BAM conversion for efficient re-analysis
Methylation Callers
BSBolt
BiSulfite Bolt is a bisulfite sequencing analysis platform that provides accurate and fast bisulfite sequencing alignments and methylation calls. It outperforms Bismark, BSSeeker2, BISCUIT, and BWA-Meth based on alignment accuracy and methylation calling accuracy.
Key advantages:
- Fast, accurate bisulfite-aware alignment
- Supports both directional and undirectional libraries
- CGmap and BedGraph output formats
- Cross-sample matrix aggregation for comparative analysis
Rastair
Rastair is an integrated software toolkit for simultaneous SNP detection and methylation calling from mC→T sequencing data (such as TAPS+ and Illumina's 5-Base chemistries). It combines machine-learning-based variant detection with genotype-aware methylation estimation.
Key advantages:
- F1 scores exceeding 0.99 for datasets above 30x depth
- Processes a 30x depth file in under 30 minutes with 32 CPU cores
- Integrated variant and methylation calling
- Standard-compliant outputs in VCF, BAM, and BED formats
- Identifies positions where variants disrupt or create CpG sites
Benchmark
BSBolt Performance
According to the BiSulfite Bolt publication, BSBolt outperforms existing bisulfite alignment tools:
| Tool | Alignment Accuracy | Methylation Calling |
|---|---|---|
| BSBolt | Highest | Most accurate |
| Bismark | Lower | Good |
| BSSeeker2 | Lower | Good |
| BISCUIT | Moderate | Moderate |
| BWA-Meth | Lower | Lower |
Rastair Performance
According to the Rastair publication on NA12878 benchmark datasets:
- F1 Score: >0.99 for datasets above 30x depth
- Processing Time: <30 minutes for 30x depth file with 32 CPU cores
- GPU Acceleration: ~2x faster with GPU available
- Additional Detection: Reports ~500,000 additional positions where SNPs create "de-novo" CpGs
Quick Start
Configuration for Primary Use Case
Default configuration uses:
- BSBolt as methylation caller (set
taps: truefor Rastair) - GRCh38 reference genome
- FASTP quality filtering
- Automatic deduplication
- Per-cytosine methylation reports
pixi run nextflow run main.nf -profile docker -resume
Prepare a Samplesheet
Create a CSV samplesheet with your input. The pipeline supports three input modes:
- FASTQ Input (Full Pipeline)
sample,lane,fastq_1,fastq_2
sample1,L001,/path/to/sample1_R1.fastq.gz,/path/to/sample1_R2.fastq.gz
sample2,L001,/path/to/sample2_R1.fastq.gz,/path/to/sample2_R2.fastq.gz
- BAM Input (Skip Alignment)
sample,lane,bam,bai
sample1,L001,/path/to/sample1.bam,/path/to/sample1.bam.bai
- CRAM Input (Skip Alignment + Auto-Convert)
sample,lane,cram,crai
sample1,L001,/path/to/sample1.cram,/path/to/sample1.cram.crai
CRAM Benefits:
- Compressed input: CRAM files are ~4x smaller than BAM
- Faster pipeline: Skip alignment step when re-running methylation calling
- Automatic conversion: CRAM→BAM conversion integrated into pipeline
Samplesheet Columns:
sample: Sample identifierlane: Sequencing lane (optional, defaults to L001)- FASTQ mode:
fastq_1,fastq_2(gzipped FASTQ files) - BAM mode:
bam,bai(aligned BAM + index) - CRAM mode:
cram,crai(compressed alignment + index)
Run the Pipeline
It will automatically detect the input format (FASTQ, BAM, or CRAM) to run the appropriate steps:
nextflow run main.nf \
--input samplesheet.csv \
--profile docker \
-resume
Advanced Options
# Run with Rastair (TAPS-based methylation)
nextflow run main.nf \
--input samplesheet.csv \
--taps true \
--profile docker \
-resume
# Run with custom trim parameters (Rastair)
nextflow run main.nf \
--input samplesheet.csv \
--taps true \
--trim_OT 10 \
--trim_OB 10 \
--profile docker \
-resume
# BSBolt with pre-built index
nextflow run main.nf \
--input samplesheet.csv \
--bsbolt_index /path/to/bsbolt_index \
--profile docker \
-resume
# Custom reference genome
nextflow run main.nf \
--input samplesheet.csv \
--reference /path/to/reference.fa \
--profile docker \
-resume
For test mode with sample data:
nextflow run main.nf -profile docker,test -resume
View Results
Output files will be generated in the results/ directory. File structure depends on pipeline mode:
-BSBolt Results:
results/alignment/*.bam- Aligned BAM filesresults/bsbolt/methylation_calls/*.cgmap.gz- CGmap format methylation callsresults/bsbolt/methylation_calls/*.bedGraph.gz- BedGraph format for visualizationresults/bsbolt/aggregate_matrix/*_matrix.txt- Cross-sample methylation matrixresults/deduplicated/*.bam- Deduplicated BAM files
-Rastair Results:
results/rastair/mbias/- M-bias calculation results and plotsresults/rastair/call/*.txt- Methylation callsresults/rastair/methylkit/*.methylkit.txt.gz- MethylKit format for R analysis
-Common output files:
results/multiqc_report.html- Interactive quality control reportresults/pipeline_info/- Execution timeline and trace logs
For more advanced usage and configuration options, see the Pipeline Architecture documentation.
References
- BiSulfite Bolt: A bisulfite sequencing analysis platform: GigaScience paper on BSBolt
- Rastair: an integrated variant and methylation caller: BioRxiv preprint on Rastair
- Pipeline Olympics: benchmarking computational workflows for DNA methylation sequencing: NAR paper on benchmarking methylation pipelines
- https://github.com/gianglabs/nf-short-read-methylation: The production-grade pipeline for short-read methylation analysis using Nextflow
Table of Contents
Recent Articles

Data Curation and Harmonization for Cancer Genomics Cohorts
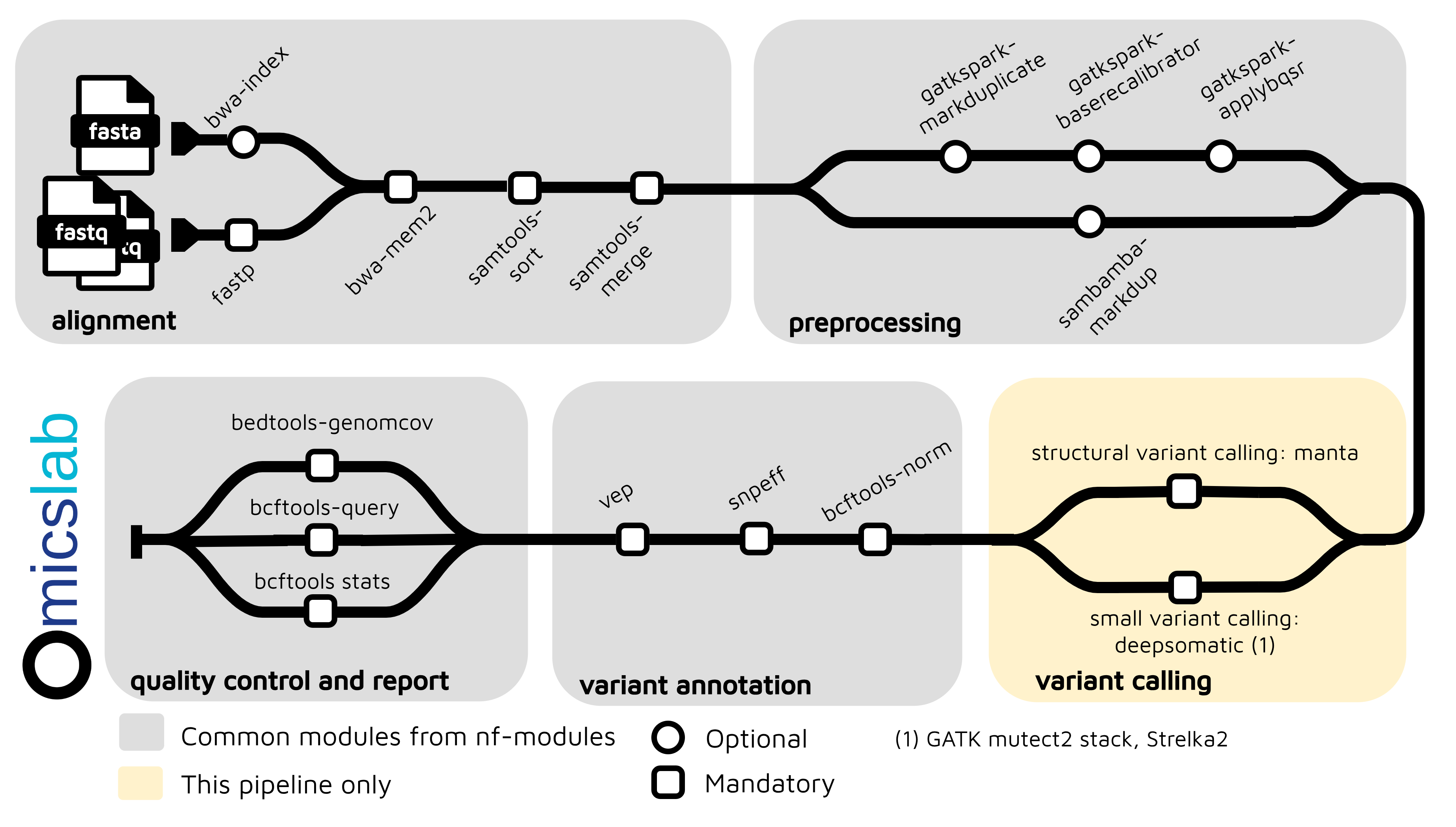
Short-read Somatic Variant Calling Pipeline Using Nextflow
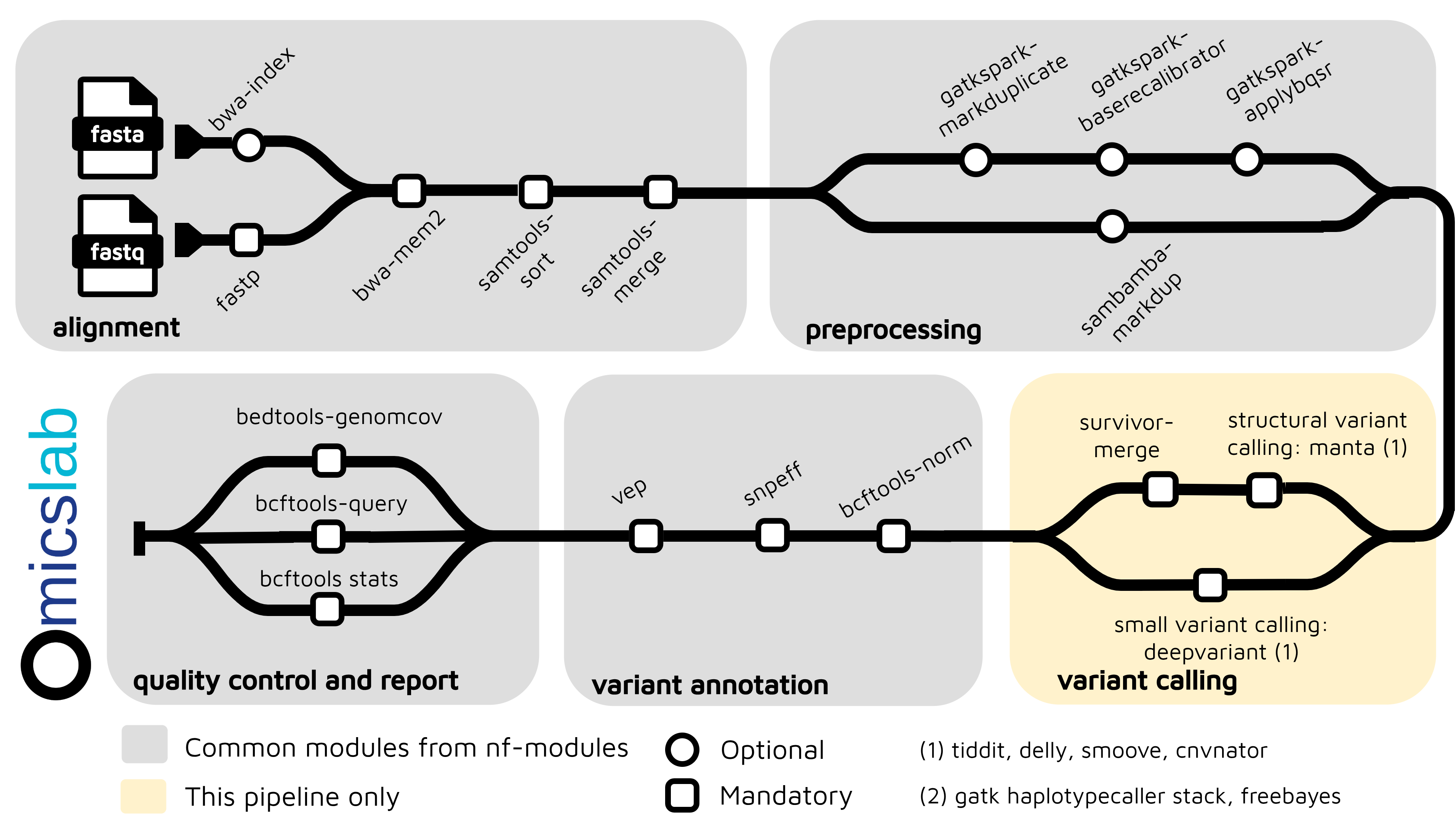
Short-read Germline Variant Calling Pipeline Using Nextflow